





Mentre negli ultimi decenni si è osservata una diminuzione della mortalità per diversi tumori, quelli del fegato e dei dotti biliari continuano a mostrare tassi di mortalità in crescita (1,2); la sopravvivenza a cinque anni per l’epatocarcinoma (HCC) è del 18% ed è seconda solo al tumore del pancreas (3).
La trasformazione fibro-nodulare del fegato è il fattore di rischio più importante; nel 90% dei casi l’HCC si sviluppa in pazienti cirrotici e la sua incidenza annuale in tali pazienti è del 2-4% (1).
Il virus dell’epatite B (HBV) ed il virus dell’epatite C (HCV) sono associati a oltre il 70% di tutti gli HCC a livello globale (4); anche il virus dell’epatite D (HDV) sicuramente gioca un ruolo nello sviluppo dell’HCC, sebbene non sia ancora stato definitivamente accertato se esso sia direttamente oncogenico (5).
Unicità dell’HDV e decorso dell’epatite D
Lo studio del ruolo dell’HDV nell’HCC è complesso a causa delle sue peculiari caratteristiche biologiche (6). Si tratta di un minuscolo virus ad RNA che usa come rivestimento l’antigene di superficie dell’epatite B (HBsAg) per formare, rilasciare e trasmettere il virione (6), mentre la sua replicazione è del tutto autonoma da quella dell’HBV (7); la simbiosi obbligata con l’HBV rende difficile studiare il ruolo specifico dell’HDV nella patogenesi tumorale.
Fin dai primi studi dell’infezione, è emerso che l’HDV è altamente patogeno ed è la causa della forma meno comune ma più grave e progressiva di epatite, l’epatite cronica D (ECD), che porta alla cirrosi epatica in un’alta percentuale (fino all’80%) dei casi. Una volta instauratasi, la cirrosi si complica in molti pazienti con lo scompenso epatico o l’HCC, accompagnati da un alto tasso di mortalità (8).
Tuttavia, non sono disponibili ampi studi prospettici sull’ECD e la maggior parte dei dati sulla storia naturale della malattia è stata dedotta da studi retrospettivi di follow-up o da studi trasversali.
L’HDV rappresenta un fattore di rischio per lo sviluppo dell’HCC?
Numerosi studi clinici in Europa, per lo più di natura retrospettiva, hanno dimostrato che l’ECD è caratterizzata da un aumentato rischio di sviluppo dell’HCC rispetto alla monoinfezione da HBV (9); l’aumento del rischio di HCC è stato confermato sia in uno studio retrospettivo in più di 2000 veterani negli Stati Uniti (10), dove è stato documentato che l’HCC poteva anche essere indipendente dalla presenza di cirrosi e direttamente oncogenico, che in uno studio svedese in cui è stato osservato che l’incidenza di HCC aumentava tra i pazienti con HBV/HDV coinfetti rispetto ai pazienti HBV monoinfetti (11).
Il ruolo oncogenico dell’HDV è stato ulteriormente confermato da una recente metanalisi di Alfaiate e coll. (12), in cui è emersa una associazione tra ECD e HCC dall’analisi di 93 lavori scientifici che comprendevano 68 studi caso-controllo e 25 studi di coorte (12). Nonostante l’eterogeneità degli studi, la metanalisi ha ribadito che i pazienti HBV/HDV coinfetti avevano un rischio significativamente più elevato di sviluppare l’HCC rispetto a quelli con monoinfezione da HBV monoinfetti, rischio che risultava ancora più evidente quando l’analisi era limitata agli studi più attendibili condotti dopo il 2010.
Lo studio di Alfaiate ha anche dimostrato un aumentato rischio di HCC dopo l’esclusione di una coinfezione da HCV, noto fattore di rischio per l’HCC spesso presente nei pazienti con HDV (12). Un aumento significativo nello sviluppo di HCC è stato osservato anche nei pazienti HDV/HIV-1 coinfetti, dove l’HDV sembra essere uno dei principali fattori che causano il tumore epatico probabilmente favorito dalla concomitante immunodeficienza (12).
Livelli di replicazione dell’HDV e dell’HBV nell’HCC associato all’HDV
Sebbene sia stata documentata una correlazione tra i livelli di replicazione dell’HDV e il rischio di complicanze epatiche (13,14) i dati disponibili sui livelli di replicazione dell’HDV e dell’HBV nei pazienti con HCC associato a HDV sono scarsi.
Recentemente, abbiamo studiato i livelli di replicazione dell’HDV all’interno del tumore analizzando il tessuto tumorale e non tumorale circostante di cinque pazienti con HCC associato a HDV. I risultati sono stati confrontati con i livelli di replicazione dell’HDV in pazienti sottoposti a trapianto di fegato per cirrosi epatica terminale da HDV senza HCC (15).
In due di cinque pazienti affetti da HDV-HCC si è osservato un netto calo dell’HDV-RNA all’interno del tumore; il calo dell’HDV-RNA si verificava tra la periferia del tumore e l’area perilesionale, mentre in tutte le aree non tumorali circostanti i livelli virali erano simili. Nei tre restanti pazienti, i livelli di RNA virale nel tessuto tumorale e non tumorale erano simili.
Pertanto, questo studio ha dimostrato che il modello di replicazione dell’HDV all’interno del tumore non è lo stesso in tutti i casi e che la ridotta replicazione virale all’interno del tumore non si sarebbe potuta rilevare testando solo i livelli di viremia dell’HDV che erano simili tra cirrosi con HCC e senza HCC; questi dati suggeriscono dunque che l’espressione dell’HDV nel tumore possa variare notevolmente.
Analizzando il DNA dell’HBV intraepatico nel tessuto tumorale e nelle aree circostanti non tumorali dello stesso fegato è stato osservato che i livelli di HBV DNA erano estremamente bassi o non rilevabili sia nel siero che nel fegato di tutti i pazienti con HDV-HCC (15) nonché nei fegati cirrotici HDV senza HCC, in linea con i bassi livelli di replicazione dell’HBV tipici dell’ECD (16); questi dati dimostrano dunque che l’HCC-HDV non è associato ad un aumento della replicazione dell’HBV.
HDV ed epatocarcinogenesi
L’epatocarcinogenesi è stata associata ad alterazioni complesse delle vie di segnalazione molecolare (17) ma i dati attualmente disponibili sui potenziali meccanismi oncogenici dell’HDV sono scarsi. Per la dipendenza obbligata dell’HDV dall’HBV, non è noto se l’HCC sia il risultato di un effetto oncogenico cumulativo dell’HBV e dell’HDV, di un effetto della cirrosi presente nel 90% dei casi di HCC, ovvero di un effetto oncogenico diretto dell’HDV.
Gli ostacoli maggiori nel definire il ruolo dell’HDV nella patogenesi dell’HCC sono i limiti dei sistemi sperimentali attualmente disponibili e le difficoltà nell’ottenere campioni di fegato da pazienti con HCC associato all’HDV, in particolare campioni di tessuto tumorale e di tessuto non tumorale circostante.
Profilo molecolare dell’HCC-HDV
L’introduzione delle tecnologie di genomica ha fornito strumenti importanti per analizzare la patogenesi di malattie complesse in cui migliaia di geni sono simultaneamente disregolati (18). Tuttavia i dati sul profilo genomico dell’HDV o sui livelli di replicazione dell’HDV all’interno del tumore sono ancora limitati.
Grazie alla disponibilità di una raccolta unica di campioni di fegato di pazienti con cirrosi HDV ed HCC e con cirrosi HDV senza HCC, tutti sottoposti a trapianto di fegato per HCC o malattia epatica terminale, è stato possibile studiare per la prima volta il profilo molecolare di questo tumore (15). Poiché il fegato contiene una popolazione cellulare eterogenea comprendente epatociti e cellule non parenchimali, il profilo dell’espressione genica è stato analizzato utilizzando epatociti isolati dal tessuto epatico tumorale e non tumorale di pazienti con HDV-HCC e tessuto epatico di pazienti con cirrosi HDV senza HCC.
I profili di espressione genica dell’HCC associato all’HDV sono stati confrontati anche con quelli di soggetti con HCC associato alla monoinfezione da HBV. Nell’HCC associato all’HDV gli epatociti maligni erano caratterizzati da un arricchimento di geni coinvolti nella regolazione del ciclo replicativo cellulare e soprattutto nel danno e riparazione del DNA, come documentato dalla presenza di sei pathways con la maggioranza dei geni upregolati (Sonic Hedgehog, GADD45, DNA-damage-induced 14-3-3s, cyclins and cell cycle regulation, cell cycle: G2–M DNA-damage checkpoint regulation, and hereditary breast cancer signaling) (15) (Figura 1).
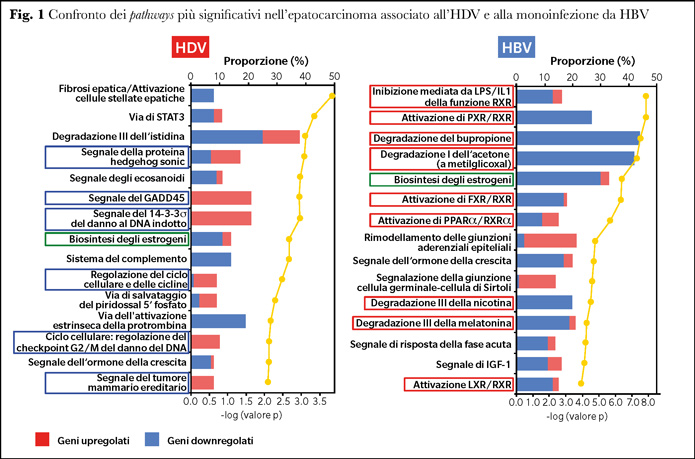
Il profilo molecolare dell’HDV-HCC è stato confrontato con quello degli epatociti maligni derivati da pazienti con monoinfezione da HBV. Nessuno dei sei pathways coinvolti nel ciclo cellulare/replicazione del DNA, danno e riparazione del DNA rilevati nell’HDV -HCC è stato identificato negli epatociti maligni dell’HCC associato all’HBV mentre il profilo molecolare dell’HDV-HCC era associato specificamente ad un arricchimento di questi geni upregolati. Pertanto, nonostante la dipendenza dell’HDV dall’HBV, il profilo molecolare dell’HDV-HCC è marcatamente diverso da quello dell’HBV-HCC.
I geni degli epatociti maligni dell’HBV erano principalmente associati ad una downregolazione dei processi metabolici, dei recettori dell’acido retinoico, del rimodellamento cellulare e delle funzioni di motilità (19) (Figura 1).
A differenza dell’HCC associato all’HBV, in cui il segno distintivo era uno spegnimento del metabolismo, il profilo genico dell’HDV-HCC ha identificato l’instabilità genomica come un potenziale meccanismo di epatocarcinogenesi dell’HDV, suggerendo che HDV e HBV promuovono la carcinogenesi attraverso meccanismi molecolari distinti (15,19).
Il panorama genomico dell’HCC, incluso l’HDV-HCC, è stato recentemente descritto in uno studio condotto in Mongolia (20), che ha la più alta incidenza di HCC nel mondo (21).
Il profilo genomico derivato da 76 pazienti con HCC associato a HBV, HCV e HDV (20), ha mostrato mutazioni comuni già note, come TP53 e CTNNB1, ma anche una serie di geni unici dell’infezione da HDV, in particolare il gene SPTA1. Questi dati suggeriscono l’esistenza di nuovi meccanismi molecolari che potrebbero svolgere un ruolo particolare nell’epatocarcinogenesi in Mongolia (20).
Conclusioni
Sebbene l’HDV non sia ancora stato incluso nella lista dei virus cancerogeni nella specie umana (5), le evidenze finora accumulate suggeriscono che il rischio di sviluppare l’HCC è maggiore nei pazienti con epatite cronica D rispetto a quelli con monoinfezione da HBV. L’HDV si replica nel nucleo degli epatociti e, attraverso la sua capacità di interagire con diverse proteine dell’ospite e di modulare la loro espressione, può alterare molteplici vie di segnalazione cellulare coinvolte nell’infiammazione, nello stress ossidativo, nell’apoptosi e nella proliferazione cellulare.
Il primo profilo molecolare definito in pazienti con HCC-HDV, nei quali l’espressione genica è stata studiata in epatociti maligni e non maligni microdissezionati (15), ha dimostrato che l’HCC associato all’HDV è caratterizzato dalla upregolazione dei geni coinvolti nel controllo della replicazione cellulare/ciclo/DNA e dal danno e riparazione del DNA, un dato che identifica l’instabilità genomica come un importante potenziale meccanismo di epatocarcinogenesi da HDV.
Poiché questo profilo genomico era peculiare dell’HDV e distinto da quello dell’HCC associato all’HBV, sembra evidente che questi due virus promuovono l’epatocarcinogenesi attraverso meccanismi diversi.
Un altro studio recente, che ha descritto il panorama genomico in Mongolia (20), ha aggiunto ulteriori informazioni sulle caratteristiche molecolari dell’HDV-HCC. Tuttavia, questi studi sono stati condotti su pochi pazienti ed è pertanto necessario estenderli a più numerose casistiche multicentriche al fine di raccogliere dati più conclusivi.
- Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359-86.
- Ryerson AB, Eheman CR, Altekruse SF, et al. Annual Report to the Nation on the Status of Cancer, 1975-2012, featuring the increasing incidence of liver cancer. Cancer 2016;122:1312-1337.
- Jemal A, Ward EM, Johnson CJ, et al. Annual Report to the Nation on the Status of Cancer, 1975-2014, Featuring Survival. J Natl Cancer Inst. 2017;109(9).
- James SL, Abate D, Abate KH, et al. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018;392:1789-1858.
- Bouvard V, Baan R, Straif K, et al. A review of human carcinogens-Part B: Biological agents. Lancet Oncol. 2009; 10(4):321-2.
- Rizzetto, M. The delta agent. Hepatology 1983;3:729-737.
- Taylor JM, Purcell RH, Farci P. Hepatitis D (Delta) Virus. In Fields Virology, 6th ed.; Knipe, D.M., MaH, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013.
- Rosina F, Conoscitore P, Cuppone R, et al. Changing pattern of chronic hepatitis D in Southern Europe. Gastroenterology 1999:117:161-166.
- Fattovich G, Giustina G, Christensen E, et al. Influence of hepatitis delta virus infection on morbidity and mortality in compensated cirrhosis type B. The European Concerted Action on Viral Hepatitis (Eurohep). Gut 2000;46:420-426.
- Kushner T, Serper M, Kaplan DE. Delta hepatitis within the Veterans Affairs medical system in the United States: Prevalence, risk factors, and outcomes. J. Hepatol. 2015;63:586-592.
- Ji J, Sundquist K, Sundquist J. A population-based study of hepatitis D virus as potential risk factor for hepatocellular carcinoma. J. Natl. Cancer Inst. 2012;104:790-792.
- Alfaiate D, Clement S, Gomes D, et al. Chronic hepatitis D and hepatocellular carcinoma: A systematic review and meta-analysis of observational studies. J. Hepatol. 2020;73:533-539.
- Niro GA, Smedile A, Ippolito AM. et al. Outcome of chronic delta hepatitis in Italy: A long-term cohort study. J. Hepatol. 2010;53:834-840.
- Kamal H, Westman G, Falconer K, et al. Long-Term Study of Hepatitis Delta Virus Infection at Secondary Care Centers: The Impact of Viremia on Liver-Related Outcomes. Hepatology 2020;72:1177-1190.
- Diaz G, Engle RE, Tice A, et al. Molecular Signature and Mechanisms of Hepatitis D Virus-Associated Hepatocellular Carcinoma. Mol. Cancer Res. 2018;16:1406-1419.
- Farci P, Niro GA. Clinical features of hepatitis D. Semin. Liver Dis. 2012;32;228-236.
- El-Serag HB. Translational research: The study of community effectiveness in digestive and liver disorders. Gastroenterology 2007;132:8-10.
- Schena M, Shalon D, Davis RW, Brown PO. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 1995;270:467-70.
- Melis M, Diaz G. Kleiner DE, Zamboni F, et al. Viral expression and molecular profiling in liver tissue versus microdissected hepatocytes in hepatitis B virus-associated hepatocellular carcinoma. J. Transl. Med. 2014;12: 230.
- Candia J, Bayarsaikhan E, Tandon, M, et al. The genomic landscape of Mongolian hepatocellular carcinoma. Nat. Commun. 2020;11:4383.
- Znaor A, Chimed T, Laversanne M, et al. The public health challenge of liver cancer in Mongolia. Lancet Gastroenterol. Hepatol. 2018;3:660-662.
